Fonctionnement d'un échographe : origine, processus et application clinique
Actuellement, l'appareil à ultrasons s'est imposée comme un outil fondamental dans le domaine de l'éducation et de la formation. imagerie diagnostique. Ce dispositif utilise le technologie des ultrasons pour obtenir des images précises et en temps réel des structures internes du corps humain, facilitant ainsi l'évaluation des organes, des tissus et des vaisseaux sanguins. sans avoir recours à des procédures invasives.
La capacité de l'échographe à fournir des informations détaillées, sûres et rapides a révolutionné la pratique clinique. L'utilisation de cet équipement médical permet aux professionnels de la santé de détecter et surveiller un large éventail de pathologies de manière précoce et efficace. En outre, sa polyvalence et sa portabilité ont permis d'étendre son utilisation à de nombreuses spécialités médicales.
L'utilisation de l'appareil à ultrasons permet de réaliser échographies dans le but de analyser les organes et les tissus au niveau interne. Il s'agit de l'une des techniques médicales les plus utilisées, car c'est une méthode rapide, efficace et non invasive. Elle est principalement utilisée pour détecter des maladies et des anomalies, pour surveiller la santé des patients, pour étudier le développement et la croissance du bébé pendant la grossesse, ainsi que pour guider certaines procédures médicales.
Contrairement à d'autres techniques d'imagerie, telles que le Rayons X ou le Tomographie axiale informatisée (CT)échographie n'utilise pas de radiations ionisantes, ce qui en fait un une technique plus sûre. En outre, son portabilité et facilité d'utilisation a permis son intégration dans les consultations, les urgences et les unités de soins intensifs, facilitant la prise de décision clinique en temps réel et améliorant les soins aux patients.
Dans l'article suivant, nous examinerons l'origine de cet équipement médical jusqu'à nos jours, le fonctionnement d'un échographe, ainsi que ses applications dans la pratique clinique.
Origine de l'échographe : des débuts à nos jours
Le développement de l'échographe est étroitement lié au développement de la technologie des ultrasons et à son application dans le domaine médical.

Premières études : découverte de l'effet piézoélectrique
Les premières études sur les ondes ultrasoniques remontent à la fin du 19e siècle, lorsque les physiciens français Pierre et Jacques Curie ont découvert, en 1880, les ondes ultrasoniques. l'effet piézoélectrique. Ce phénomène physique est la capacité de certains matériaux, comme le quartz et certains cristaux de céramique, à générer une charge électrique lorsqu'ils sont soumis à une pression mécanique.
L'importance de l'effet piézoélectrique dans les ultrasons est fondamentale, car il constitue la base de l'échographie. les bases du fonctionnement des transducteurs/sondes à ultrasons. En pratique, les cristaux piézoélectriques du transducteur convertissent les signaux électriques en vibrations ultrasoniques (ondes ultrasoniques), qui sont transmises au corps du patient. Par effet piézoélectrique, ces échos sont transformés en signaux électriques qui sont traités par l'appareil à ultrasons pour générer des images en temps réel.
Développement du premier échographe : de l'effet piézoélectrique et des ultrasons au domaine médical.
Suite à la découverte de l'effet piézoélectrique, le phénomène a d'abord été appliqué dans les domaines industriel et militaire, notamment dans le domaine de l'énergie. le développement de sonars pour la détection sous-marine pendant la Première et la Seconde Guerre mondiale.
Cependant, le potentiel des ultrasons et de l'effet piézoélectrique pour générer et recevoir des ondes acoustiques n'est pas passé inaperçu dans la communauté scientifique et médicale. Les l'adaptation de cette technologie au domaine médical a débuté au milieu du 20e siècle. Le médecin écossais Ian Donald, avec l'ingénieur Tom Brownétaient les les chercheurs pionniers qui ont commencé à appliquer l'effet piézoélectrique à l'examen cliniquePremier du genre, il a jeté les bases de l'échographie médicale.
Plus précisément, c'est dans les années 1950 que des chercheurs ont mis au point le système d'information sur les maladies infectieuses. premier prototype d'échographe clinique. Dans un premier temps, l'échographe a été utilisé en obstétrique pour la visualisation du fœtus et la détection des pathologies pendant la grossesse, ce qui a constitué une révolution dans le suivi prénatal.
Des années 1960 et 1970 à nos jours : les progrès de l'échographie
Au cours des années 1960 et 1970, la technologie de l'échographie a connu des avancées significatives. Elle est devenue des images fixes aux échographies en temps réelCela a permis d'observer les le mouvement des organes et des structures internes. Par la suite, l'incorporation de la L'effet Doppler a permis d'étudier le flux sanguin et l'évaluation vasculaire.Le nouveau système d'échographie élargit encore les applications cliniques de l'échographe.
Au cours des dernières décennies, la le développement de la technologie et de l'informatique a permis l'émergence de des échographes plus compacts, portables et à plus haute résolution. Aujourd'hui, l'échographe est un outil sûr, efficace et polyvalent utilisé dans un large éventail de spécialités, de la médecine d'urgence à la cardiologie, en passant par la gynécologie et la médecine interne. À ce titre, il est devenu un équipement médical indispensable dans la pratique médicale.
Échographes de nouvelle génération : innovation, technologie et intelligence artificielle
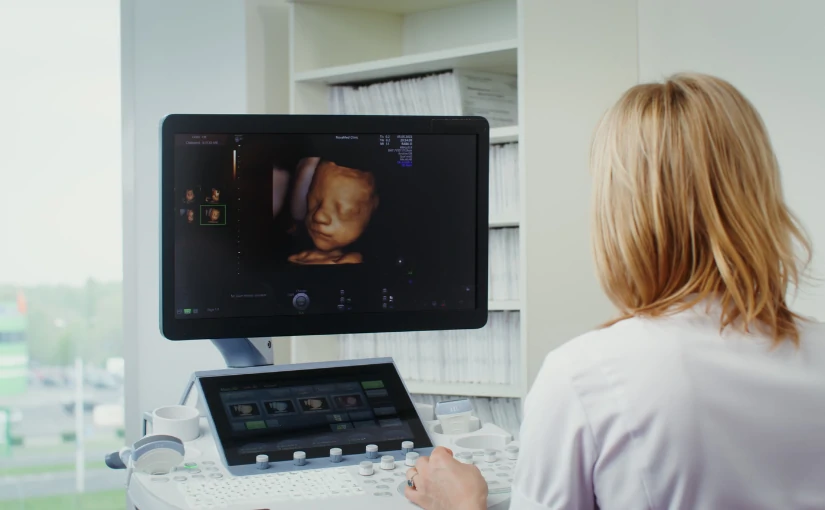
Ces dernières années, la technologie a beaucoup évolué dans le domaine de la médecine. Les des échographes de pointe offrir des images en Technologie 3D, 4D et 5Det permettent donc visualiser l'intérieur du corps humain en mouvement et en temps réel.
L'une des innovations les plus récentes concerne les échographes qui intègrent les éléments suivants systèmes de traitement numérique qui appliquent la l'intelligence artificielle dans l'analyse d'images médicales. L'utilisation d'un Logiciel d'IA dans l'équipement d'échographie permet d'améliorer la vitesse, l'efficacité, la sécurité et la précision du diagnostic, en fournissant une analyse avancée et détaillée qui améliore la prise de décision clinique.
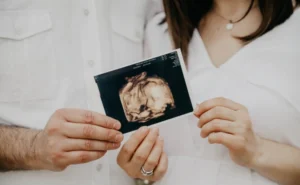
Dans ce domaine, il convient de souligner l'importance de l'action de l'Union européenne dans le domaine de l'éducation. l'utilisation d'échographes de pointe pour visualiser le fœtus en temps réel. Il est connu sous le nom de ultrasons émotionnels et permet aux parents de faire connaissance avec leur bébé avant sa naissance. Ce type d'échographie combine la Technologie 3Dqui fournit des images tridimensionnelles, avec la technologie 4D et 5DLes mouvements du bébé sont visibles en temps réel avec une clarté et une qualité d'image élevées. Il est ainsi possible de voir les principaux mouvements du bébé. Du bâillement au changement de position en passant par l'ouverture des yeux.
Comment fonctionne un appareil à ultrasons : Procédure étape par étape
Les échographes sont un outil essentiel dans la pratique médicale. Comprendre son fonctionnement et le déroulement d'un examen échographique est essentiel pour garantir la qualité du diagnostic et la sécurité des patients. Dans les pages qui suivent, nous expliquons le fonctionnement d'un échographe et la procédure étape par étape :

1. préparation du patient et application du gel conducteur
Premièrement, le patient reçoit des instructions sur la position l'endroit où il doit être positionné en fonction de la zone à scanner et du type de diagnostic à établir. Avant de commencer l'examen, le gel conducteur sur la peau du patient. Il a pour fonction d'éliminer l'air généré entre la peau de la zone à examiner et le transducteur ou la sonde à ultrasons, facilitant ainsi la transmission des ondes ultrasonores.
Sélection du type de transducteur
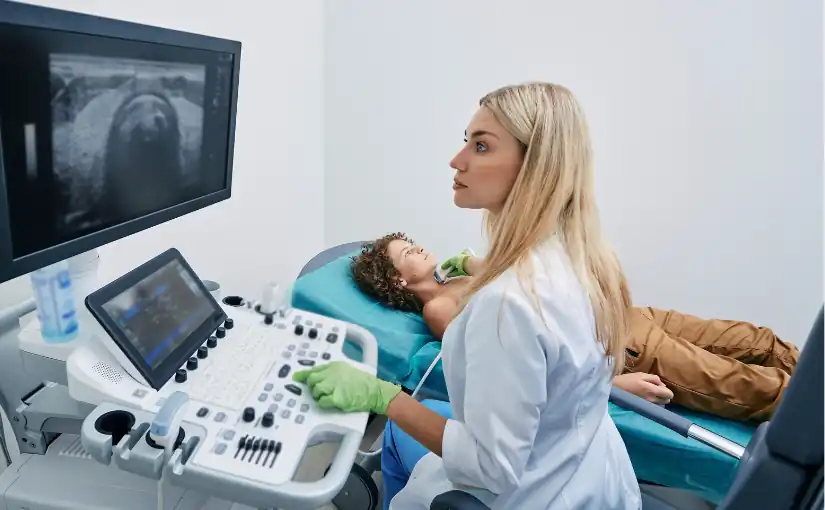
L'un des principaux composants de l'appareil à ultrasons est le transducteur ou la sonde. Il existe différents types de transducteursChacun d'entre eux est conçu pour scanner des régions et des profondeurs différentes. Alors que les transducteurs linéaires sont utilisés pour les études vasculaires et superficielles, les modèles convexes sont utiles pour les scanners abdominaux profonds.
C'est donc le professionnel de la santé qui sera chargé de sélectionner les le type de transducteur approprié, le connecter à l'équipement et vérifier son bon fonctionnement. avant de commencer l'étude.
3. Émission et réception d'ultrasons
Une fois le transducteur préparé, l'opérateur le place sur la zone recouverte de gel. Le le transducteur émet des ondes ultrasonores à haute fréquence qui pénètrent les tissus internes du patient. Lorsque ces ondes traversent le corps et sont réfléchies aux interfaces des différents tissus et organes, la les ondes réfléchies, appelées échos, reviennent vers le transducteur.
4. Ramassage de l'écho
Le transducteur fait également office de récepteuren détectant les ondes réfléchies (échos) générées par les différentes structures internes. Ces échos contiennent des informations sur la localisation et les caractéristiques des tissus traversésCela permet d'analyser l'état et le fonctionnement des différents organes.
5. Traitement de l'image
Les échos captés par le transducteur sont convertis en signaux électriques.qui sont traitées par la console d'échographie à l'aide de différents algorithmes. Le résultat est la génération d'images bidimensionnelles ou tridimensionnelles en temps réel qui sont affichées sur l'écran de l'appareil.
Grâce à l'analyse des images médicales, l'opérateur peut observer l'anatomie et le mouvement des organes et des structures internes. Le mode Doppler permet quant à lui d'étudier le flux sanguin dans les tissus.
6. Exploration systématique
Le professionnel effectue une le balayage méthodique en déplaçant le transducteur sur la zone d'intérêt à analyserL'examen est réalisé en obtenant différentes coupes (longitudinales, transversales, obliques) afin d'examiner complètement les organes et les structures. Cette exploration systématique est essentielle pour obtenir une un diagnostic complet et détailléLes résultats de l'étude doivent être documentés de manière à ce que les résultats pertinents ne soient pas omis et que les résultats soient documentés de manière adéquate.
7. Réglage des paramètres de l'image
Pendant le balayage, l'opérateur peut ajuster les différents paramètresLe mode d'affichage (2D, 3D, Doppler) peut être réglé à partir du gain (luminosité), de la profondeur et de la mise au point sur le mode d'affichage (2D, 3D, Doppler). Il est ainsi possible de optimiser la qualité de l'image et l'adapter aux caractéristiques anatomiques du patient.
8. Interprétation des images médicales
Après l'examen, le médecin est chargé de analyser les images obtenues en temps réelL'enregistrement des données, l'identification des perturbations possibles et la réalisation de captures statiques ou d'enregistrements de séquences pertinentes. Grâce à ces enregistrements, le rapport final peut être entièrement documenté, ce qui servira de base à l'élaboration du rapport final. diagnostic et les la prise de décision clinique.
9. Achèvement et nettoyage
À la fin de l'étude, l'opérateur retire le gel de la peau du patient. Par la suite, un le protocole de désinfection et de nettoyage du matériel utiliséLe transducteur et la surface de contact entre chaque patient.
Ce processus structuré fait de l'échographie une technique rapide, sûre, non invasive et très polyvalente, facilitant l'évaluation précise de multiples organes et pathologies dans la pratique clinique quotidienne.
Principales applications cliniques des ultrasons
Les échographes sont utilisés dans pratiquement toutes les spécialités médicales. Les principales applications cliniques concernent les domaines suivants :
Obstétrique et gynécologie
L'échographie est essentielle dans la suivi de la grossesseElle est utilisée pour évaluer le développement du fœtus, la localisation et la viabilité de la grossesse, la détection des anomalies congénitales et le suivi des complications obstétricales. Elle est également utilisée pour l'étude de les pathologies gynécologiquescomme des kystes ovariens, des fibromes utérins ou des anomalies de l'endomètre.
Cardiologie
Les échocardiographie est une technique essentielle pour la l'évaluation de l'anatomie et de la fonction cardiaquespermettant de diagnostiquer maladies valvulaires, cardiomyopathies, insuffisance cardiaque, maladies cardiaques congénitales et évaluer le flux sanguin en utilisant le mode Doppler.
Médecine interne et gastro-entérologie
Les échographie abdominale permet d'examiner des organes tels que le foie, la vésicule biliaire, le pancréas, les reins, la rate et la vessie. Il joue donc un rôle clé dans la le diagnostic des masses, des kystes, des calculs, des inflammations et d'autres pathologies. Il est également utilisé dans l'évaluation de l'ascite et dans la surveillance des procédures interventionnelles.
Examen vasculaire
Au moyen de l'outil Échographie Dopplerest évaluée, le le flux sanguin dans les artères et les veinesIl est donc utilisé dans le diagnostic de la thrombose veineuse profonde, de l'insuffisance veineuse, de la sténose artérielle, des anévrismes et d'autres maladies vasculaires.
Appareil locomoteur
Permet étudier les muscles, les tendons, les ligaments, les articulations et les tissus mous, faciliter le diagnostic des blessures sportives, déchirures, tendinites, bursites, hémorragies et masses sous-cutanées.
Urologie
Il est utilisé pour l'évaluation de la prostate, de la vessie, des testicules et des reinsutiles pour le diagnostic de la maladie de l'hyperplasie prostatique, la lithiase, les tumeurs et autres troubles urologiques.
Pédiatrie
L'échographie est particulièrement utile dans l'étude des pathologies pédiatriques, telles que le dysplasie de la hanche, hydrocéphalie, malformations rénales et anomalies abdominales chez les nouveau-nés et les nourrissons.
Orientations sur les procédures interventionnelles
L'échographe facilite l'exécution des tâches suivantes biopsies, drains, ponctions, pose de cathéters et autres interventionsL'utilisation de la procédure augmente la sécurité et la précision de la procédure.
Médecine d'urgence et soins intensifs
Sa rapidité et sa portabilité permettent un diagnostic immédiat des pathologies graves telles que les épanchements pleuraux, l'hémopéritoine, le pneumothorax, la tamponnade cardiaque et l'évaluation rapide des polytraumatisés (échographie FAST).
Conclusion
L'échographie s'est imposée comme un outil essentiel dans la pratique clinique.Il offre une analyse médicale précise, efficace, sûre et en temps réel des différents organes et tissus internes. Ses non invasifL'absence de radiations ionisantes et sa capacité à s'adapter à l'évolution de l'environnement. plusieurs spécialités en font une ressource indispensable tant pour l'évaluation initiale que pour le suivi de nombreuses pathologies.
Votre portabilité et rapidité faciliter la prise de décision clinique dans divers contextes, de la consultation externe aux situations d'urgence. Depuis ses origines jusqu'à aujourd'hui, l'utilisation des ultrasons a révolutionné la pratique médicale en améliorant la qualité des soins. la qualité des soins de santé et en contribuant de manière décisive à une un diagnostic précoce, précis et sûr pour les patients.
Si vous souhaitez obtenir plus d'informations sur les échographes ou d'autres équipements de diagnostic médical, vous pouvez nous contacter. Notre équipe 4D vous conseillera sur la meilleure solution pour votre clinique ou votre hôpital.
Bibliographie
Société espagnole de soins intensifs pédiatriques (SECIP) (2018). Principes de base de l'échographie. Consulté le 20 mai 2025 à l'adresse suivante https://secip.com/images/uploads/2018/09/1-FUNDAMENTOS-BASICOS-DE-ECOGRAF%C3%8DA.pdf
Authorea (n.d.). Ultrasons : principes physiques et applications cliniques. Authorea. Consulté le 20 mai 2025 à l'adresse suivante https://www.authorea.com/doi/full/10.22541/au.172660489.98960333

Administrateur de Diagximag. Distributeur d'équipements et de solutions d'imagerie médicale.
![]()